Edwards Lifesciences (NYSE: EW) today announced investments that reflect the company’s deep commitment to advancing patient care through structural heart innovation, addressing large unmet patient needs and supporting sustainable long-term growth.
Edwards has entered into an agreement to acquire JenaValve Technology, a pioneer in the transcatheter treatment of aortic regurgitation (AR), a deadly disease that impacts a significant and growing population and is largely untreated today. JenaValve presented positive results of its U.S. pivotal trial for the treatment of symptomatic, severe AR in high-risk patients late last year. As the pioneer in valve innovation for more than 60 years, Edwards believes it is uniquely positioned to lead this next frontier of aortic valve disease treatment. Edwards anticipates FDA approval of the JenaValve Trilogy Heart Valve System in late 2025, which will represent the first approved therapy for patients suffering from AR.
Building on an investment made in 2016, Edwards has exercised its option to acquire Endotronix, a leader in heart failure (HF) management solutions. Many structural heart patients Edwards serves today also suffer from HF with limited options. This acquisition will expand Edwards’ structural heart portfolio into a new therapeutic area to address the large unmet needs of patients suffering from HF. Last month, Endotronix received FDA approval for Cordella, an implantable pulmonary artery pressure sensor allowing early, targeted therapeutic intervention. A CMS national coverage determination is expected in early 2025.
“These acquisitions expand our opportunities to address the unmet needs of aortic regurgitation and heart failure patients around the world,” said Bernard Zovighian, Edwards’ CEO. “We are pleased to enter these structural heart therapeutic areas with innovation, world-class science and clinical evidence to provide access to life-saving technologies for patients around the world.”
Edwards anticipates these investments will strengthen its leadership in structural heart innovation and represent long-term growth opportunities. Edwards expects minimal revenue contribution from these acquisitions in 2025. The aggregate upfront purchase price for these strategic investments is approximately $1.2 billion. The acquisitions are subject to the satisfaction of certain closing conditions, including the receipt of required antitrust and foreign investment approvals.
About Edwards Lifesciences
Edwards Lifesciences is the global leader of patient-focused innovations for structural heart disease and critical care monitoring. We are driven by a passion for patients, dedicated to improving and enhancing lives through partnerships with clinicians and stakeholders across the global healthcare landscape. For more information, visit www.edwards.com and follow us on Facebook, Instagram, LinkedIn, X and YouTube.
This news release includes forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. We intend the forward-looking statements contained in this press release to be covered by the safe harbor provisions of such Acts. These forward-looking statements can sometimes be identified by the use of forward-looking words, such as “may,” “might,” “believe,” “will,” “expect,” “project,” “estimate,” “should,” “anticipate,” “plan,” “goal,” “continue,” “seek,” “intend,” “optimistic,” “aspire,” “confident” and other forms of these words and include, but are not limited to, statements made by Mr. Zovighian and statements regarding our expected continued performance of Edwards; performance of the Edwards, JenaValve or Endotronix technologies; product and therapy benefits; patient access and outcomes; size of treatable population; leading position; growth opportunities; unmet needs in structural heart, aortic regurgitation, and heart failure therapeutic areas; probability of approval by the FDA and in the anticipating timeline; probability of a positive NCD by the CMS and in the anticipated timeline; synergies between the technologies, business, and operations of each of JenaValve and Endotronix and Edwards’ technologies, products, portfolio, expertise, and operations; ability to leverage the technology or innovation from these acquisitions or cause or ensure accelerated access to life-saving technologies for patients or development of novel technologies as a result of these acquisitions; commitment to expand opportunities in structural heart innovation, address large unmet patient needs, and support sustainable long-term growth; objective to expand Edwards’ portfolio into new structural heart therapeutic areas; opportunities and revenue return on these acquisitions and their contribution to Edwards’ growth and performance, as well as the expectations on timing of such returns and contributions; therapy approval pipeline for patients suffering from AR; probability of the closing of the two acquisitions; other objectives and expectations; and other statements that are not historical facts. Forward-looking statements are based on estimates and assumptions made by management of the company and are believed to be reasonable, though they are inherently uncertain and difficult to predict. Our forward-looking statements speak only as of the date on which they are made, and we do not undertake any obligation to update any forward-looking statement to reflect events or circumstances after the date of the statement. Investors are cautioned not to unduly rely on such forward-looking statements.
Forward-looking statements involve risks and uncertainties that could cause actual results or experience to differ materially from that expressed or implied by the forward-looking statements. Factors that could cause actual results or experience to differ materially from that expressed or implied by the forward-looking statements include, but are not limited to: (i) Edwards may be unable to close the acquisitions of each of JenaValve and Endotronix, which may materially and adversely affect Edwards’ business and the price of Edwards’ common stock; (ii) the occurrence of any event, change or other circumstance that could cause Edwards to abandon the acquisitions of either or both of JenaValve and Endotronix; (iii) risks related to disruption of management’s attention from Edwards’ ongoing business operations; (iv) the effect of the announcement or the pendency of the acquisitions on Edwards’ relationships with its customers, operating results and business generally; (v) potential significant transaction costs associated with either or both acquisitions; (vi) the outcome of any legal proceedings or regulatory actions to the extent initiated against Edwards or others related to either or both acquisitions; (vii) the ability of Edwards to execute on its strategy and achieve its goals and other expectations after the closing of either or both acquisitions; (viii) legal, regulatory, tax and economic developments affecting Edwards’ business; (ix) the unpredictability and severity of catastrophic events, including, but not limited to, acts of terrorism, outbreak of war or hostilities or current or future pandemics or epidemics, as well as Edwards’ response to any of the aforementioned factors; and (x) other risks detailed in Edwards’ filings with the SEC, which may be found at edwards.com.
Edwards, Edwards Lifesciences, and the stylized E logo are trademarks of Edwards Lifesciences Corporation or its affiliates. All other trademarks are the property of their respective owners.
Contacts
Media Contact: Amy Hytowitz, 949-250-4009
Investor Contact: Mark Wilterding, 949-250-6826




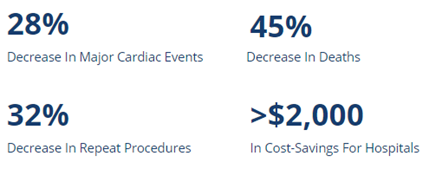

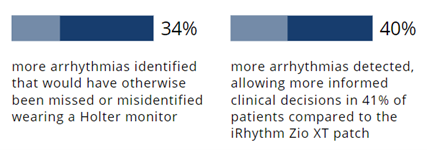
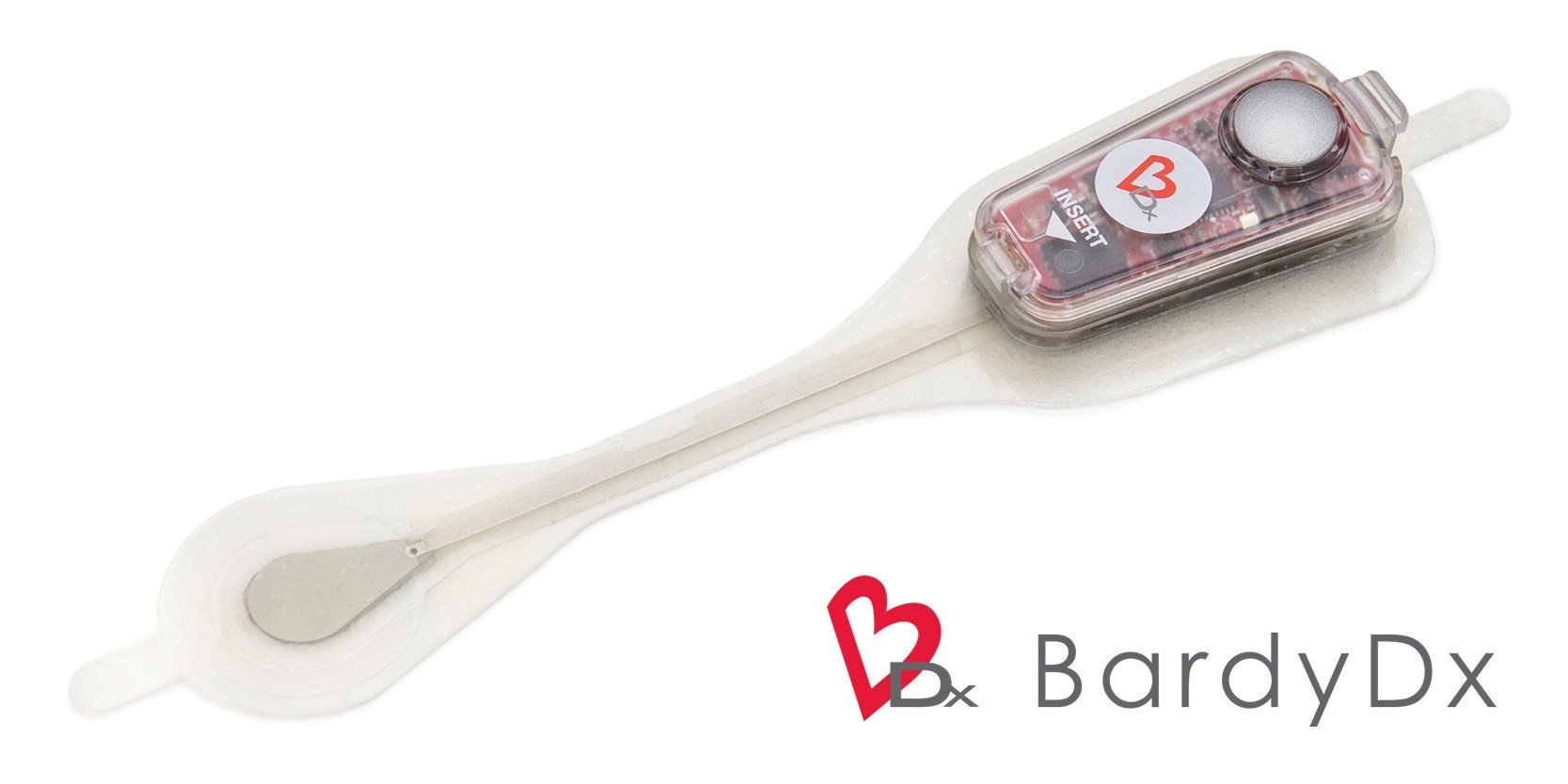 The Carnation Ambulatory Monitor (CAM™) is the first non-invasive, P-wave centric ambulatory cardiac monitor and arrhythmia detection device designed to elevate signal detection, patient compliance, and physician care procedures. Through advanced compression algorithms to more effectively process signals, and short patch vectors to overcome signal-to-noise limitations, the technology is rigorously optimized to ensure a clear P-wave recording and superior detection of heart rhythms. The form and placement of the device are designed to improve comfort for patients, while generating higher rhythm specificity. The sensor is placed over the sternum, on top of the heart, this location prevents any noisy muscle stimulation and enhances the capture of the P-wave signal.
The Carnation Ambulatory Monitor (CAM™) is the first non-invasive, P-wave centric ambulatory cardiac monitor and arrhythmia detection device designed to elevate signal detection, patient compliance, and physician care procedures. Through advanced compression algorithms to more effectively process signals, and short patch vectors to overcome signal-to-noise limitations, the technology is rigorously optimized to ensure a clear P-wave recording and superior detection of heart rhythms. The form and placement of the device are designed to improve comfort for patients, while generating higher rhythm specificity. The sensor is placed over the sternum, on top of the heart, this location prevents any noisy muscle stimulation and enhances the capture of the P-wave signal. underserved needs of comfort for the female chest. With a lifestyle-enabling form factor patients can remain going about their daily activities comfortably and confidently. CAM™ provides options for a 48-hour, 7- or 14-day wear period and continuous 24/7 monitoring using BDxCONNECT, a versatile patient management portal. When experiencing symptoms, patients simply push the button on the device, and a report is sent to their physician to analyze and determine if intervention is needed. Once the wear period is finished, the data is uploaded and a more in-depth view of ECG data is generated through multiple fields of concise analysis. In an industry-leading two-day report turn-around, physicians have an improved resolution of ECG data, supplying more heart rhythm information and allowing for the detection of many unrecognized patterns and more clinically-actionable diagnoses.
underserved needs of comfort for the female chest. With a lifestyle-enabling form factor patients can remain going about their daily activities comfortably and confidently. CAM™ provides options for a 48-hour, 7- or 14-day wear period and continuous 24/7 monitoring using BDxCONNECT, a versatile patient management portal. When experiencing symptoms, patients simply push the button on the device, and a report is sent to their physician to analyze and determine if intervention is needed. Once the wear period is finished, the data is uploaded and a more in-depth view of ECG data is generated through multiple fields of concise analysis. In an industry-leading two-day report turn-around, physicians have an improved resolution of ECG data, supplying more heart rhythm information and allowing for the detection of many unrecognized patterns and more clinically-actionable diagnoses.
 Cognitive deficits are the hallmark of a variety of psychiatric and neurodegenerative disorders affecting millions of patients worldwide. These conditions include; major depressive disorder (MDD), schizophrenia, a number of neurodegenerative disorders including Alzheimer’s Disease, and aging in general. Treatment options are few and far between and in many cases no treatment options are available to address cognitive deficits across disorders which often leave patients, care givers and family members struggling. At the forefront of research in the area is Dr Etienne Sibille and his lab at the Centre for Mental Health and Addiction (CAMH) at the University of Toronto, a world leading academic research institute. Dr. Sibille and his team has been studying how brain circuitry is affected and if there are ways in which the connections between neurons in vital areas responsible for cognition can be strengthened and as we age.
Cognitive deficits are the hallmark of a variety of psychiatric and neurodegenerative disorders affecting millions of patients worldwide. These conditions include; major depressive disorder (MDD), schizophrenia, a number of neurodegenerative disorders including Alzheimer’s Disease, and aging in general. Treatment options are few and far between and in many cases no treatment options are available to address cognitive deficits across disorders which often leave patients, care givers and family members struggling. At the forefront of research in the area is Dr Etienne Sibille and his lab at the Centre for Mental Health and Addiction (CAMH) at the University of Toronto, a world leading academic research institute. Dr. Sibille and his team has been studying how brain circuitry is affected and if there are ways in which the connections between neurons in vital areas responsible for cognition can be strengthened and as we age. Dr. Sibille’s group embarked on a journey to design a drug that could reverse the pathology that they discovered and hypothesized was leading to cognitive decline in patients. By the summer of 2017, when we first met Dr. Sibille and his associate Dr. Prevost, — they, CAMH and international collaborators had filed patents on a class of compounds that showed promise in laboratory studies to address this issue. It was easy for us to be engaged by the vision, need and opportunity. While the team had initially identified a group of novel compounds and very interesting biology, translating that into a drug was still far off. From 2017 to 2021, through hard work, tenacity, constant collaboration with other drug developers in the field, leveraging connections through various local and international accelerator programs, VC’s like us, clinicians, etc. the team was able to identify a lead drug that had all the characteristics needed. They also learned the art of approaching and working with investors, contract organizations that do much of the preclinical work, the various regulatory bodies like FDA, and clinicians experienced running clinical studies. Through this process the company settled on a program to first address the huge unmet clinical need in patients that respond to treatment of MDD using antid epressants but continue to have lingering cognitive issues. It is estimated that in the US and Europe alone that there are more than 4.3 million suffering from cognitive impairment that have been successfully treated for MDD. Therapeutic strategies currently available today include expensive psychotherapy, cognitive remediation and more recently transcranial magnetic stimulation and currently there are no approved therapeutics to treat the cognitive symptoms.
Dr. Sibille’s group embarked on a journey to design a drug that could reverse the pathology that they discovered and hypothesized was leading to cognitive decline in patients. By the summer of 2017, when we first met Dr. Sibille and his associate Dr. Prevost, — they, CAMH and international collaborators had filed patents on a class of compounds that showed promise in laboratory studies to address this issue. It was easy for us to be engaged by the vision, need and opportunity. While the team had initially identified a group of novel compounds and very interesting biology, translating that into a drug was still far off. From 2017 to 2021, through hard work, tenacity, constant collaboration with other drug developers in the field, leveraging connections through various local and international accelerator programs, VC’s like us, clinicians, etc. the team was able to identify a lead drug that had all the characteristics needed. They also learned the art of approaching and working with investors, contract organizations that do much of the preclinical work, the various regulatory bodies like FDA, and clinicians experienced running clinical studies. Through this process the company settled on a program to first address the huge unmet clinical need in patients that respond to treatment of MDD using antid epressants but continue to have lingering cognitive issues. It is estimated that in the US and Europe alone that there are more than 4.3 million suffering from cognitive impairment that have been successfully treated for MDD. Therapeutic strategies currently available today include expensive psychotherapy, cognitive remediation and more recently transcranial magnetic stimulation and currently there are no approved therapeutics to treat the cognitive symptoms.



